Ø
Goals of Classification
A
classification system should have two qualities.
a.
Stability
b.
Predictability
Stability- Frequent changes in the classification
system can cause confusion. Therefore, it is important to strive for the
development of a classification system that requires only minor adjustments
when new information becomes available. This will help minimize confusion and
ensure stability in the classification system.
Predictability-
By
knowing the characteristics of one member of a taxonomic group, it should be
possible to assume that the other members of the same group probably have similar
characteristics.
Predictability
is an important characteristic of a classification system. It refers to the
ability to anticipate and understand how the system categorizes and organizes
information. A predictable classification system follows consistent rules and
criteria, allowing users to have a clear understanding of how entities are
classified and how they relate to each other.
Ø
General Methods of Classification
1.
Intuitive method
2.
Phenetic or Phenotypic classification
3.
Phylogenetic or Phyletic classification
4.
Genotypic or Genetic classification
5.
Numerical taxonomy or Adansonian
classification
In
this method, a microbiologist who has been studying the properties of the
organisms for several years may decide that the organisms under study represent
one or more species or genera. The drawback of this system is that the
characters considered important by one person may not be so important to others.
So different taxonomists may arrive at very different groupings. This is a primitive
method that is not in use now. However,
some schemes based on intuitive methods have proved to be useful.
2.
Phenotypic
classification
For
a very long time, microbial taxonomists relied on this system. This classification
system is based on the mutual similarity in the phenotypic or morphological
characteristics of the organisms. This system succeeded in bringing order to
biological diversity. To some extent, this system also clarified the function associated
with morphological structures. For eg: Flagella and motility are associated in
most organisms. So, it could be assumed that flagella are involved in at least
some types of motility.
Phenetic
studies can also reveal evolutionary relationships. However, this system is not
dependent on phylogenetic analysis. The phenetic system is based purely on
morphological characters. This system will compare as many traits as possible, giving
equal weightage to all traits studied. The best phenetic classification is the
one constructed by comparing as many attributes/ morphological characters as
possible. Organisms sharing many characteristics (>70%) are grouped into a
single taxon.
3.
Phylogenetic or Phyletic
classification
With
the publication of Darwin’s “On the Origin of Species’ in 1859, biologists
began to develop phylogenetic or phyletic classification system. This
classification system is based on the evolutionary relationships of organisms. The
term “phylogeny” means ‘evolutionary development of a species’. In Greek, ‘phylon’
means ‘tribe or race’ and ‘genesis’ means ‘origin or generation.’ However, for
much of the 20th century, microbiologists could not effectively
employ phylogenetic classification systems mainly due to the lack of good
fossil records. When Carl Woese and George E Fox proposed the use of
rRNA nucleotide sequences to assess evolutionary relationships among
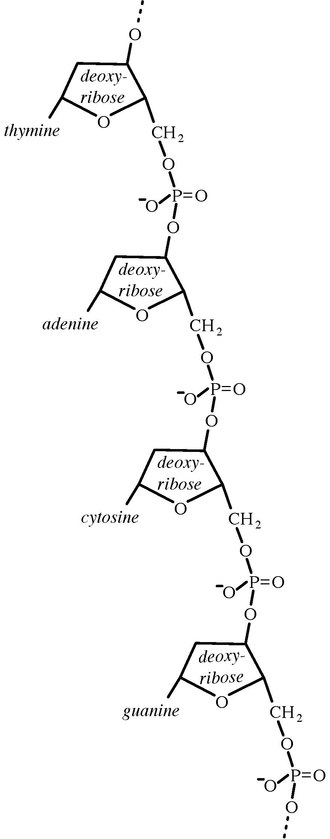
microorganisms, the doors opened for phylogenetic classification systems. 16S ribosomal RNA (or 16S rRNA)
is the RNA component of the 30S subunit
of a prokaryotic ribosome (SSU rRNA).
It binds to the Shine-Dalgarno sequence and provides most of the SSU
structure. The genes coding for it are referred to as 16S rRNA gene and
are used in reconstructing phylogenies, due to the
slow rates of evolution of
this region of the gene. The validity of this approach is widely accepted and
over 2 lakh different 16S and 18S rRNA sequences are saved in the International
databases- GenBank and the
Ribosomal Database Project (RDP-II).
4.
Genotypic or Genetic
classification
In
the genotypic classification system, genetic similarity between the organisms is
evaluated. In this method comparison of either the individual gene or whole
genome (haploid set of chromosomes in a microorganism) is done. Since 1970, it
is widely accepted that if the genome of prokaryotes shows similarity in more
than 70%, they could belong to the same species. Microbial genomes could be
compared in many ways. The simplest method is DNA Base Composition
determination. Other ways include Nucleic acid hybridization, Nucleic acid
sequencing, and Genomic fingerprinting (evaluation of genes that evolve more
quickly than those that encode rRNA.
5.
Numerical taxonomy or
Adansonian classification
Numerical
taxonomy, also known as numerical phenetics or phenetic taxonomy, is a method
used in biological classification to group organisms based on their overall
similarity with the help of computers. Unlike traditional taxonomy, which
relies on morphological characteristics and evolutionary relationships,
numerical taxonomy employs quantitative data from various attributes or
characteristics of organisms. This approach aims to create classifications
based on the overall similarity or dissimilarity of organisms rather than their
shared ancestry.
The
process of numerical taxonomy involves several steps:
1.
Data Collection: Quantitative data is collected from the organisms under study.
This data can include various traits such as morphological features,
biochemical characteristics, physiological measurements, or genetic markers.
2.
Data Standardization: The collected data is standardized to ensure
compatibility and comparability across different organisms. This step may
involve converting measurements to standardized units or transforming data to
eliminate biases or variations.
3.
Similarity or Dissimilarity Calculation: A similarity or dissimilarity matrix
is constructed based on the standardized data. Different mathematical methods
can be used to calculate the degree of similarity or dissimilarity between
pairs of organisms. Common methods include the Jaccard coefficient or
correlation coefficients.
4.
Cluster Analysis: Cluster analysis is performed using the similarity or
dissimilarity matrix to group organisms into clusters or taxa.
5.
Taxonomic Hierarchy: The clusters obtained from the cluster analysis are
organized into a taxonomic hierarchy. This hierarchy can include higher-level
groupings, such as classes or orders, as well as lower-level groupings, such as
genera or species.
Numerical
taxonomy has been widely used in fields such as ecology, microbiology, botany,
and zoology. It provides a systematic and quantitative approach to classify
organisms based on overall similarities, without relying on subjective
interpretations or expert judgments. However, it is important to note that
numerical taxonomy does not consider evolutionary relationships explicitly,
which can be a limitation in certain contexts where phylogenetic information is
crucial for understanding the evolutionary history of organisms.